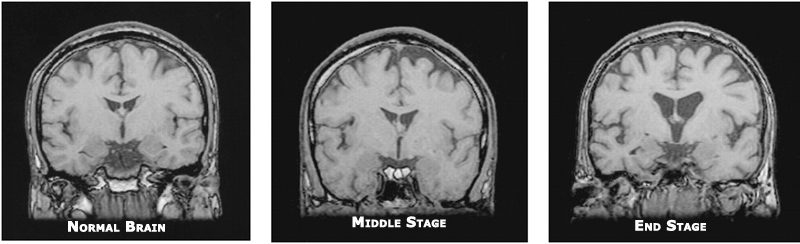
Corticobasal degeneration (CBD) – also known as corticobasal ganglionic degeneration (CBGD) – is a rare (occurs in less than 1% of the population) and progressive form of dementia.
The onset of symptoms typically occurs after the age of 60 and the average duration of the disease from onset of symptoms to death is six years.
Although the underlying cause of CBD is unknown, what is known is that CBD is the result of extensive and severe damage in multiple areas of the brain.
Research into this form of dementia is relatively new (it was discovered in 1968), but the most current research has found that there are similar, but not identical, changes in the brain protein tau to the changes observed in progressive supranuclear palsy and Pick’s Disease.
 These areas of the brain where damage is extensive include the cortex (especially in the frontal lobe and parietal lobes) and the deep-brain basal ganglia region of the brain, with the hallmark feature in that area being significant neuron degeneration and the loss of pigment in dopaminergic neurons (signifying a decrease in dopamine production) in the substantia nigra, which controls movement.
These areas of the brain where damage is extensive include the cortex (especially in the frontal lobe and parietal lobes) and the deep-brain basal ganglia region of the brain, with the hallmark feature in that area being significant neuron degeneration and the loss of pigment in dopaminergic neurons (signifying a decrease in dopamine production) in the substantia nigra, which controls movement.
Dopamine is a chemical produced by the brain (a neurotransmitter) that plays a leading role in movement, memory, pleasure, cognition, behavior, attention, sleep, and mood.
 When dopamine production decreases in the substantia nigra, movement is severely affected.
When dopamine production decreases in the substantia nigra, movement is severely affected.
Often this is the first visible symptom of CBD. It presents as stiff movement, shaky movement, jerky movement, slow movement, and increased lack of balance, increased lack of coordination, and clumsiness. Generally, movement problems affect one side of the body almost exclusively, but as CBD progresses, both sides of the body are affected.
Since these movement disorders can mimic both Parkinson’s Disease and the effects of a deep-brain stroke – one of the classic movement disorders associated with these is ideomotor apraxia (a common example is the inability to initiate walking where the foot seems to be stuck to the floor and can’t be lifted spontaneously to take a step forward) – those must be ruled out as the causes of the movement disorders.
Other early symptoms of CBD can include difficulty controlling the mouth muscles, cognition problems, and behavioral problems. Language and speech difficulties – dysphasia (an impaired ability to understand or use the spoken word) and dysarithia (an impaired ability to clearly articulate the spoken word) – are also early CBD symptoms.
(In my latest book, You Oughta Know: Recognizing, Acknowledging, and Responding to the Steps in the Journey Through Dementias and Alzheimer’s Disease, I devote a whole chapter to a comprehensive and in-depth discussion of the communication problems, including the different types of dysphasia, that occur with dementias and Alzheimer’s Disease, and ways to work with our loved ones to keep the lines of communication open for as long as possible.)
It is not unusual for CBD to be initially diagnosed, if the first symptoms are cognitive impairment and/or behavioral issues, as Alzheimer’s Disease or frontotemporal dementia. Similarly, if movement disorders are the first symptoms, CBD is often initially diagnosed as Parkinson’s Disease.
However, a clear diagnosis of CBD is usually made when both movement disorders and cognitive impairment and/or behavior problems appear simultaneously.
There is no known treatment for CBD. Unlike Parkinson’s Disease where dopamine-enhancing or dopamine-mimicking medications prove to be effective for some of the duration of the disease, these drugs have proven to be ineffective for treating CBD (this is likely because of the very different pathologies in the development and progression of the two diseases).
In the early stages of CBD, speech therapy and physical therapy may help with communication and stiffness and movement. However, as the disease progresses, these will become less effective and, in the end stage, they will be completely ineffective.
As CBD progresses, other symptoms appear and worsen, including:
- Rigidity
- Tremors
- Involuntary muscle contractions
- Involuntary eyelid spasms
- Loss of sensory functions
- “Alien hand/limb” syndrome (hand or limb movement that the person isn’t aware of nor has control over)
Because of the increased rigidity and lack of muscle coordination and use as CBD progresses, usually within five years of onset, sufferers will be unable to swallow and will be completely immobile. Even before this, though, one of the potentially-fatal risks associated with CDB is aspiration of food into the lungs because of impaired swallowing and the high likelihood of pneumonia as a result.
While a feeding tube may be considered as an alternative when CBD has progressed to the point where swallowing is significantly affected, it is, in my opinion, inhumane because it only prolongs the suffering from a disease that is ultimately fatal.
This is a quality-of-life choice. I can’t imagine for myself a life prolonged where I am completely immobile and completely dependent on everyone else for everything and I can do nothing for myself.
A feeding tube would be my worst nightmare. And for me, it would be the most cruel thing those in charge of making medical decisions for me could do to me.
Fortunately, I already have all my documents in place to make sure this can’t and won’t happen to me when and if the time comes that the choice needs to be made, because I’ve already made the choices.
So, as an aside, I would strongly urge everyone who reads this to get your wishes formalized and signed and communicated so that you have control over the end game of your life in this area.
Not only is the wise and prudent thing to do, but it eliminates the agony of wondering what to do so often seen in families where the person affected never talked about what he or she wanted and never took the time to answer these questions when he or she could.