There are several types of common early-onset dementias. Early-onset dementias are categorized as dementias where the onset of symptoms is prior to age 65. These dementias can occur as early as the 30’s, but more typically become symptomatic in the 40’s and 50’s.
Early-onset dementias, unfortunately, are still off the main grid for medical staff – a classic instance of fixed expectations that dementias won’t be an issue for a person until after age 65 – and our loved who are diagnosed with early-on dementias face challenges that our older loved ones who are suffering with these diseases don’t face. These include:
- Difficulty getting a correct diagnosis
- Loss of employment and income
- Difficulty getting Social Security Disability Insurance, Medicaid, and other employment-related disability insurance
- Loss of health insurance and high-out-of-pocket costs for medical care
- High out-of-pocket costs for long-term care
- Lack of appropriate medical care, residential care, and community services (all of these are geared toward an older population)
Early-onset dementias typically are harder to diagnose because other than the dementia systems, sufferers are usually healthy, active, and aware there is a problem.
Additionally, the symptoms of early-onset dementias usually don’t have memory impairment as the predominant feature. Most often, behavioral and personality changes occur first, so usually the first type of treatment is psychiatric instead of neurological.
The causes of early onset-dementias fall into three categories: random, genetic, and lifestyle.
Random early-onset dementias are just that. There’s no concrete link to a cause. My opinion is that few of these in this category are actually random, but the causative issue(s) have not been identified yet.
Genetics plays an important role in certain early-onset dementias (and, although the scientific community has overlooked or disregarded the familial aspect of elder-onset dementias, it appears very likely, from observation, that if there’s a family history of elder-onset dementias, there may be a genetic predisposition for development of elder-onset dementias in subsequent generations).
Three genes are known to have mutations in the case of some sufferers of early-onset dementia, Alzheimer’s type (symptoms related to these genetic mutations usually begin in the 30’s and 40’s):
- Amyloid precursor protein gene (APP) on chromosome 21
- Presenillin-1 (PSEN-1) on chromosome 14
- Presenillin-2 (PSEN-2) on chromosome 1
We’ve talked extensively here about lifestyle dementias with regard to management of health (blood pressure and blood sugar) and substance abuse, as well as with regard to what we eat and how we live daily life. Some of the early-onset dementias we will talk about here can be directly attributed to lifestyle.
There are several types of early-onset dementias.
At least 1/3 of all sufferers diagnosis with early-onset dementia have Alzheimer’s Disease (remember that Alzheimer’s Disease is a type of dementia, but is not inclusive of all types of dementia, just as all photocopiers are not Xerox photocopiers and all facial tissues are not Kleenex facial tissues).
Onset symptoms include progressive and episodic memory loss, as well as visuospatial and perceptual deficiencies, but intact language and social functioning.
 This type of early-onset dementia is more common in women than men. Once symptoms appear, the duration of the disease averages eight years.
This type of early-onset dementia is more common in women than men. Once symptoms appear, the duration of the disease averages eight years.
A recent example is the 2011 diagnosis of former University of Tennessee women’s head basketball coach, Pat Summit, who was diagnosed with early-onset dementia, Alzheimer’s type, at age 59. Coach Summitt stayed with the team one more season, but was not actively coaching that season.
Coach Summitt retired in 2012 and has begun the Pat Summit Foundation to raise Alzheimer’s Disease awareness.
 The novel, Still Alice, written by neuroscientist Lisa Genova, gives a scientific, compassionate and compelling look from the inside out of a 50-year-old Harvard psychology professor as early-onset dementia, Alzheimer’s type enters and progresses through her life.
The novel, Still Alice, written by neuroscientist Lisa Genova, gives a scientific, compassionate and compelling look from the inside out of a 50-year-old Harvard psychology professor as early-onset dementia, Alzheimer’s type enters and progresses through her life.
Since the publication of Still Alice in 2007, Genova has continued her work with bringing the neuroscience of all types of dementias in the same compassionate and compelling style of her first novel through subsequent books and through documentaries produced with her husband, who is a filmmaker.
 The second most common type of early-onset dementia is vascular dementia. Vascular dementia can occur because of:
The second most common type of early-onset dementia is vascular dementia. Vascular dementia can occur because of:
- Multiple cortical infarcts (small areas of tissue that have died from the lack a blood supply) that are most often caused by transient ischemic attacks (TIA’s) or silent strokes and characterized by stepwise deterioration of cognitive function
- Small-vessel disease, resulting in a more subtle decline of cognitive function
- Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)
- Rare cause of early-onset subcortical strokes and dementia
- Caused by a mutation of Notch 3 gene on chromosome 19
- MRI shows diffuse white matter lesions on the cerebral hemispheres, especially in the anterior temporal lobes and external capsules
With early-onset vascular dementia, there are usually lifestyle factors involved such as uncontrolled or undetected high blood pressure and an unhealthy diet. Recent scientific research has also linked high cholesterol levels with the development of vascular dementia.
 The third most common type of early-onset dementia is frontotemperal dementia (FTD), also known as Pick’s Disease, which affects the frontal and temporal lobes of the brain. FTD usually has an onset between the ages of 45 and 65. Its average duration is eight years.
The third most common type of early-onset dementia is frontotemperal dementia (FTD), also known as Pick’s Disease, which affects the frontal and temporal lobes of the brain. FTD usually has an onset between the ages of 45 and 65. Its average duration is eight years.
There are three types of FTD: behavioral variant FTD, semantic dementia, and primary progressive (also known as progressive nonfluent) aphasia.
In about half the cases of FTD, there is a positive family history for the disease, indicating a probable genetic link (although researchers have not yet identified the genetic mutation).
FTD can co-occur with motor neuron diseases (ALS, also known as Lou Gerhig’s Disease, is an example of a motor neuron disease), but only about 10% of sufferers of only motor neuron diseases develop dementia, resulting in a very aggressive course of the illness.
FTD presents differently from early-onset Alzheimer’s Disease and early-onset vascular dementia because the first symptoms involve changes in personality and social conduct while memory, perception, and visuospatial skills remain unchanged.
The most common indicators are:
- Behavior disturbances
- Personality changes
- Decreased motivation
- Reduced empathy
- Impaired planning
- Impaired judgment
- Speech and language difficulties
As FTD progresses, other symptoms become apparent:
- Difficulty behaving appropriately in new and unfamiliar situations
- Loss in inhibitions (disrobing is not uncommon)
- Loss of social skills
- Emotional outbursts
- Impulsivity
- Executive function deficits
- Decreased verbal fluency
- Compulsive or repetitive behavior
- Lack of insight
- Self-neglect
- Inappropriate sexual behavior
The semantic dementia form of FTD includes symptoms of:
- Difficulty with correctly naming objects (people, places, and things)
- Impaired understanding of the meaning of words
- Inability to understand substitute words
However, in this form of FTD, speech remains fluent and cognition remains intact. MRI scans show more atrophy of the anterior temporal lobe than the posterior temporal lobe.
The primary progressive (progressive nonfluent) aphasia form of FTD is characterized by:
- Progressive decline in all language skills, with no other cognitive deficits
- Increased difficulty with speech and speaking (by the end of the disease, most sufferers don’t speak at all)
- Speech and speaking is not fluent and requires a great deal of effort
MRI scans show predominant atrophy of the left perisylvian region of the temporal lobe.
The fourth most common type of early-onset dementia is Lewy Body dementia. I’ve included the link to my post about Lewy Body dementia for a full description, but will include a brief summary of the dementia’s 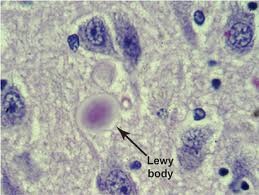 primary symptoms:
primary symptoms:
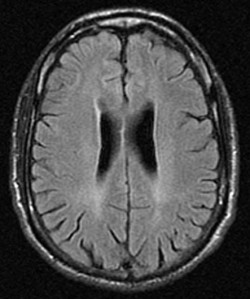
The fifth most common type of early-onset dementia is Wernicke-Korsakoff Syndrome (alcohol-related dementia). This is a lifestyle dementia, brought on by long-term, heavy alcohol consumption.

Characteristics of Wernicke-Korsakoff Syndrome include:
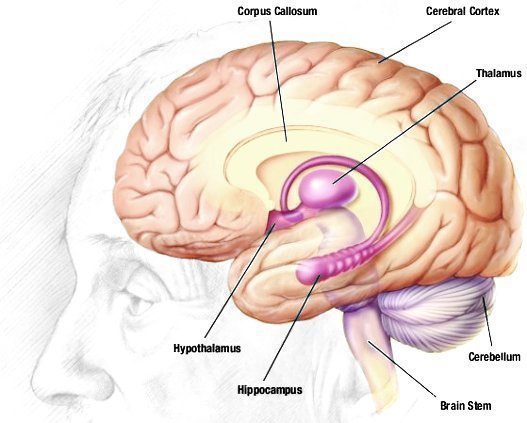
- Damage to the limbic structures and frontal lobes
- Memory impairment
- Executive functioning impairment
- Autobiographical memory is frequently affected resulting in confabulation (making up stories)
- Memory loss stops where it is when drinking stops (damage already done remains)
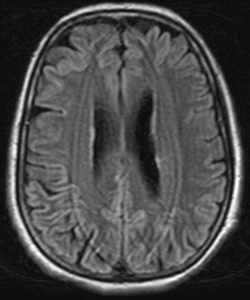
As shown by the MRI scan above, there is general cortical atrophy along with damage to the frontal, parietal and cingulated regions of the brain, with the majority of the damage occurring in the frontal lobe.
There are two other less common types of early-onset dementia that we’ll discuss.
One is Huntington’s Disease. As this genetically-inherited disease progresses, dementia develops.
 Everyone is born with this gene. However, in Huntington’s Disease, an inherited mutated copy of this gene (on chromosome 4), produces a defective form of the huntingtin protein that causes degeneration and death of the neurons, especially in the center of the brain.
Everyone is born with this gene. However, in Huntington’s Disease, an inherited mutated copy of this gene (on chromosome 4), produces a defective form of the huntingtin protein that causes degeneration and death of the neurons, especially in the center of the brain.
Because this gene is a dominant gene (as opposed to a recessive gene), everyone who inherits the mutated copy of the gene will, at some point, develop Huntington’s Disease.
Symptoms typically appear between ages 30 and 50, but it can begin at a very young age or appear in the very elderly. Primary symptoms include:
- Lack of muscle coordination in the arms, legs, head, face and upper body
- Progressive decline in thinking and reasoning skills, including memory, concentration, judgment and the ability to plan and organize
- Mood disturbances, including depression, anxiety, anger, and irritability
- Obsessive-compulsive behaviors
The last type of early-onset dementia, which is extremely rare, is Creutzfeldt-Jakob Disease (CJD). CJD is characterized by rapid neurological degeneration. It is always fatal, and death usually occurs within six months to a year of onset.
CJD belongs to a class of human and animal diseases called transmissible spongiform encephalopathies (TSEs), because the infected brain looks like a sponge. The average age of onset for CJD is 60.
“Mad Cow Disease” is the bovine equivalent of CJD (although it tends to affect younger people, with the average age of onset being 26).
There are three types of CJD:
- Sporadic (no known cause) – accounts for about 90% of cases
- Inherited (family history of the disease) – accounts for 5-10% of cases
- Acquired (transmitted by exposure to brain or nervous system tissue, usually through certain medical procedure) – accounts for less than 1% of cases
 The symptoms of CJD include:
The symptoms of CJD include:
- Rapidly progressive dementia
- Problems with muscular coordination
- Personality changes, including impaired memory, judgment, and thinking
- Impaired vision
- Insomnia
- Depression
- Lethargy
As CJD progresses, mental impairment becomes severe. Sufferers often develop involuntary muscle jerks (myoclonus), and they may go blind.
Eventually, they lose the ability to move and speak and become comatose. Pneumonia and other infections often occur as well, and they generally end in death.